Biophysical Analysis
Biophysical analyses give insight into the compound-target-interaction on a molecular level. The way a compound acts on the target shapes the drug’s PK/PD profile and efficacy in vivo. Therefore, in a targeted drug discovery approach, knowledge of the molecular behavior of your lead candidates early in the drug discovery process aids modeling the structure-activity relationship for optimal modifications of the drug design.
Our Reaction Oncology Platform enables the investigation of various aspects of the mechanism of action of a new drug, including:
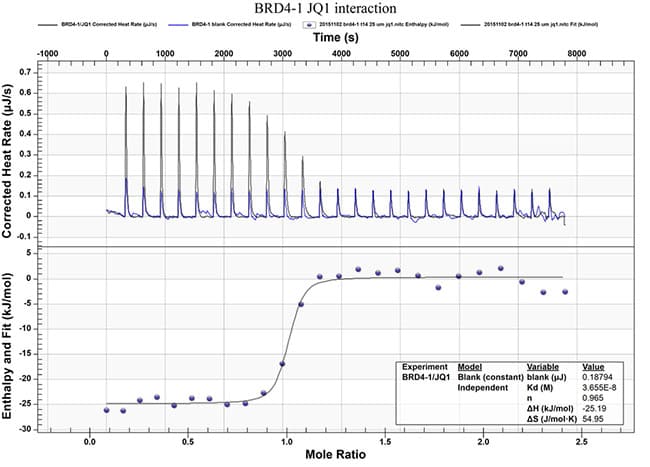
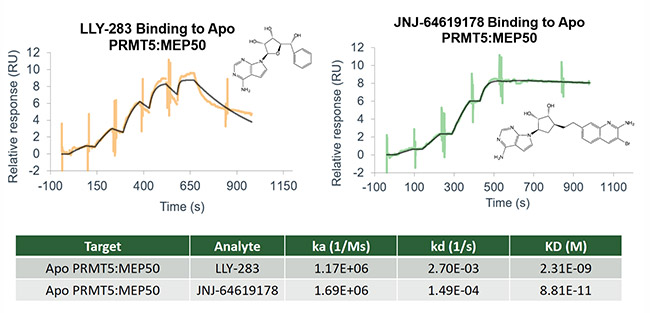
- Binding affinity
- Binding kinetics and residence time
- Target Occupancy
- Binding Reversibility
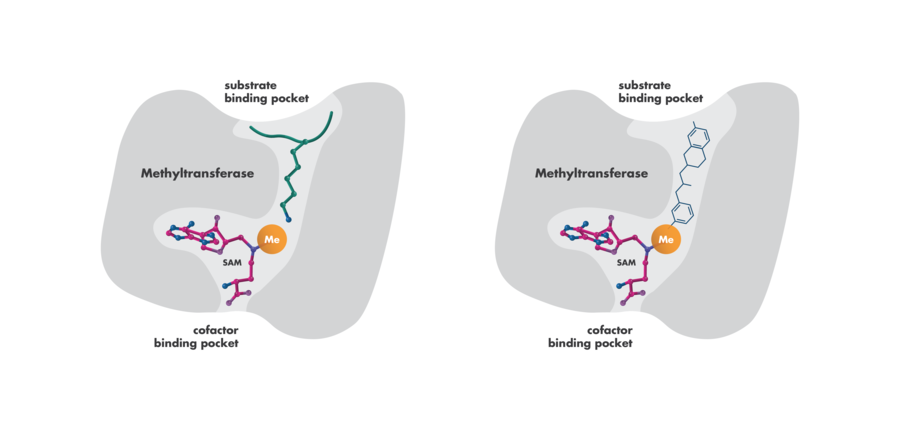
- Competition with Substrates or Co-factors
- Stoichiometry
- Thermodynamics of the compound-target interaction
The knowledge of the compound-target interaction can be expanded by examination of the structure of the compound-target complex via NMR and X-ray crystallography via an external partner.