Biophysical characterization of compounds disrupting the SARS-CoV-2 Spike – ACE2 interaction using SPR
Without rock-solid biophysical predictors of binding affinity and kinetics, the road from molecular screening and simulation to cell-based assays and in vivo testing is virtually unnavigable. Fully characterizing your COVID-19 drug candidate with a proven SPR methodology is the fundament of the successful development of efficacious drugs.
Association and dissociation constants give insight into an inhibitor’s potential efficacy and safety
The determination of the equilibrium dissociation constant, KD, for binding of a potential drug to a target is the first step for the identification of hits. For therapeutic options to prevent SARS-CoV-2 entry into host cells, the drug might either block the spike protein or the ACE2 receptor. The KD is an essential experimental triage parameter used to narrow down potential drug candidates initially identified by docking studies and molecular dynamics simulations.
To get a deeper picture of how a drug candidate might actually perform clinically – in other words, some insight into its potential efficacy and safety – the KD needs to be accurately dissected into its more useful components, the association, and dissociation kinetic constants kon and koff. These constants relate to the equilibrium dissociation constant according to KD= koff/kon. The reason for this is that equilibrium kinetics describe a closed system where the drug and target are held at constant concentrations over time.
Researchers need to know these on-off rates in order to feed more sophisticated models of what would happen when people take the drug. Getting good data upfront can only help downstream pharmacokinetic and pharmacodynamic investigations. The former, specified as concentration vs. time, captures what the body does to the drug (including rates at which it is metabolized and eliminated), while the latter, specified as effect vs. time, seeks to define what the drug does to the body.
In an actual tissue microenvironment, where association rate often dominates over diffusion rate, and targets or receptors can be locally abundant, then significant rebinding effects are expected to occur. A fast kon can therefore significantly increase the duration of target occupancy even when the koff is faster than the elimination rate. In any realistic micro-pharmacokinetic accounting of the situation, there will likely be more than one target present and so kinetic selection becomes important. The association rate to the main target relative to any secondary targets is what frequently determines clinical efficacy and safety. In a nutshell, kon is often a better predictor of side effects than koff.
Disruption of SARS-CoV-2 Spike-ACE2 interaction by compounds can be investigated by Surface Plasmon Resonance
The technique by which other techniques have historically benchmarked their binding and kinetic data is Surface Plasmon Resonance (SPR). There are many good reasons for this. For one, binding affinity can be sensitively determined kinetically, by obtaining the koff and kon, and also at steady state by plotting the maximum responses (on rate = off rate) at different concentrations, and then taking the concentration for a half-maximal response.
One of the most useful assays for an efficient Spike receptor binding domain (RBD) – ACE2 drug discovery pipeline is the SARS-CoV-2 Spike – ACE2 disruption assay. In addition to the stand-alone kinetic data, researchers want to see how their compounds perform when both Spike RBD and ACE2 are part of the assay. In other words, how the compound affects the interaction of Spike RBD and ACE2, and what concentrations of the compound are required to get the desired effect.
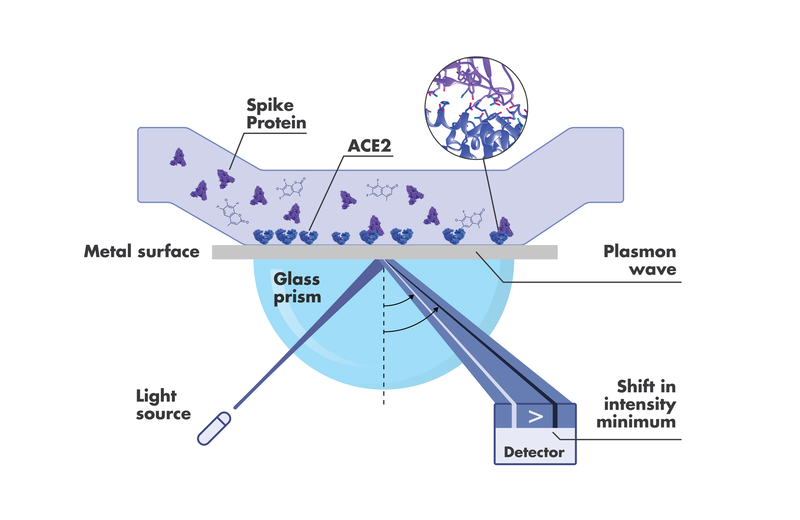
If one is looking to target the ACE2 side of the equation with a drug, for example, one important question would be whether or not binding affects the normal enzymatic ACE2 activity. Orthogonal screening via protease activity assays before moving on to cell-based or other activity analysis can give the researcher more confidence in their drug candidate selections. Finding the best inhibitor obviously entails a lot more than choosing the one with the strongest, fastest, or longest binding capacity. Being able to derive potential indicators for off-target effects or PK/PD profiles, right out of the gate, can help ensure the drug discovery process lands with the safest and most effective candidates.
Frequently Asked Questions
Q: What is the typical workflow for disruption assays for drug discovery screening drugs blocking the interaction of SARS-CoV-2 Spike and ACE2 receptor?
The workflow for the disruption assays is to:
- Establish the conditions for attaching ACE2 or spike RBD to the chip to measure their binding interactions.
- Measure the binding between ACE2 and Spike RBD in the absence of a test molecule to determine the kinetic parameters (on/off-rates and KD).
- We then measure the binding response of ACE2 or Spike RBD (whichever is not on the chip) to the target in the absence of test molecule.
- Then we flow the test molecule over the target to allow it to pre-bind before we flow the test molecule plus ACE2 or Spike RBD. This step is repeated with increasing concentrations of the test molecule.
- We then compare the binding response for the Spike RBD – ACE2 interaction in the absence and presence of the test molecule to see if there is a reduction in binding response.
Q: What is the timeline for testing substances in the Spike RBD/ACE2 disruption assay?
With each of our three Biacore 8K instruments, we can test 8 substances in parallel. For providing full concentration-response curves plus assay validation and QC we can test about 16 test molecules in one week with one machine. For simple binding studies with either ACE2 or Spike RBD, we have a higher turnover which multiplies when screening in a single concentration for hit identification.
Q: Is there a good reference inhibitor to use for either binding studies with compound/spike, compound/ACE2, or disruption of spike/ACE2 interaction?
We use MLN-4760 as a reference/control molecule for compound/ACE2 binding. Currently, there are not a lot of small molecules that have been unambiguously shown to bind Spike with 1:1 stoichiometry that is commercially available. The few antibodies or peptides in the literature that are able to block the ACE2/Spike interaction, are still mostly unavailable commercially.
Q: Should I start my SPR investigation with the disruption assay to measure the blocking of Spike/ACE2 interaction or first run a pre-screen to determine the best binder to my target?
We would recommend running a direct binding assay first. That way an appropriate concentration range (a range in which binding was observed for the direct binding assay) can be tested during the disruption assay. There is the possibility the “best” binder (in terms of KD from the direct binding assay) may not be the best at blocking the Spike/ACE2 interaction depending on where it binds, the impact on the protein structure, etc. So it may still be worth pursuing more than just the analyte with the tightest KD.
Q: What is the glycosylation status of the proteins that you use?
Spike RBD and ACE2 protein were expressed in human cell culture and thus exhibit glycosylation which ensures a high translatability to proteins in humans.
Q: Is it possible to screen spike RBD with a mutation in your assay?
We will validate the binding kinetics of the Spike/ACE2 each time we work with a new construct. Thus, there will be development period and fee before the screening of the test molecule can be done.
Q: Is the SPR approach limited to certain substances?
We can screen a variety of molecules: small molecules, peptides, proteins, antibodies, DNA/RNA, etc. There are considerations for each in the assay set-up, and we may need to make adjustments. If we need more large-scale changes to our standard assay set-up we may need to book the work as an assay development followed by testing.
Related Resources

